

1 INDICATIONS AND USAGE
Semaglutide is indicated as an adjunct to diet and exercise to improve glycemic control in adults with type 2 diabetes mellitus.
Limitations of Use
Semaglutide is not indicated for use in patients with type 1 diabetes mellitus.
2 DOSAGE AND ADMINISTRATION
2.1 Overview of semaglutide Formulations
- Use only one kind of semaglutide formulation; do not use both formulations at the same time.
- Do not take more than one tablet per day.
2.2 Important Administration Instructions
- Take semaglutide on an empty stomach in the morning with water (up to 4 ounces of water). Do not take semaglutide with other liquids besides water.
- After taking semaglutide, wait at least 30 minutes before eating food, drinking beverages, or taking other oral medications.
- Swallow tablets whole. Do not split crush, or chew.
- If a dose is missed, skip the missed dose, and take the next dose the following day.
2.3 Recommended Starting, Escalation, and Maintenance Dosage of semaglutide
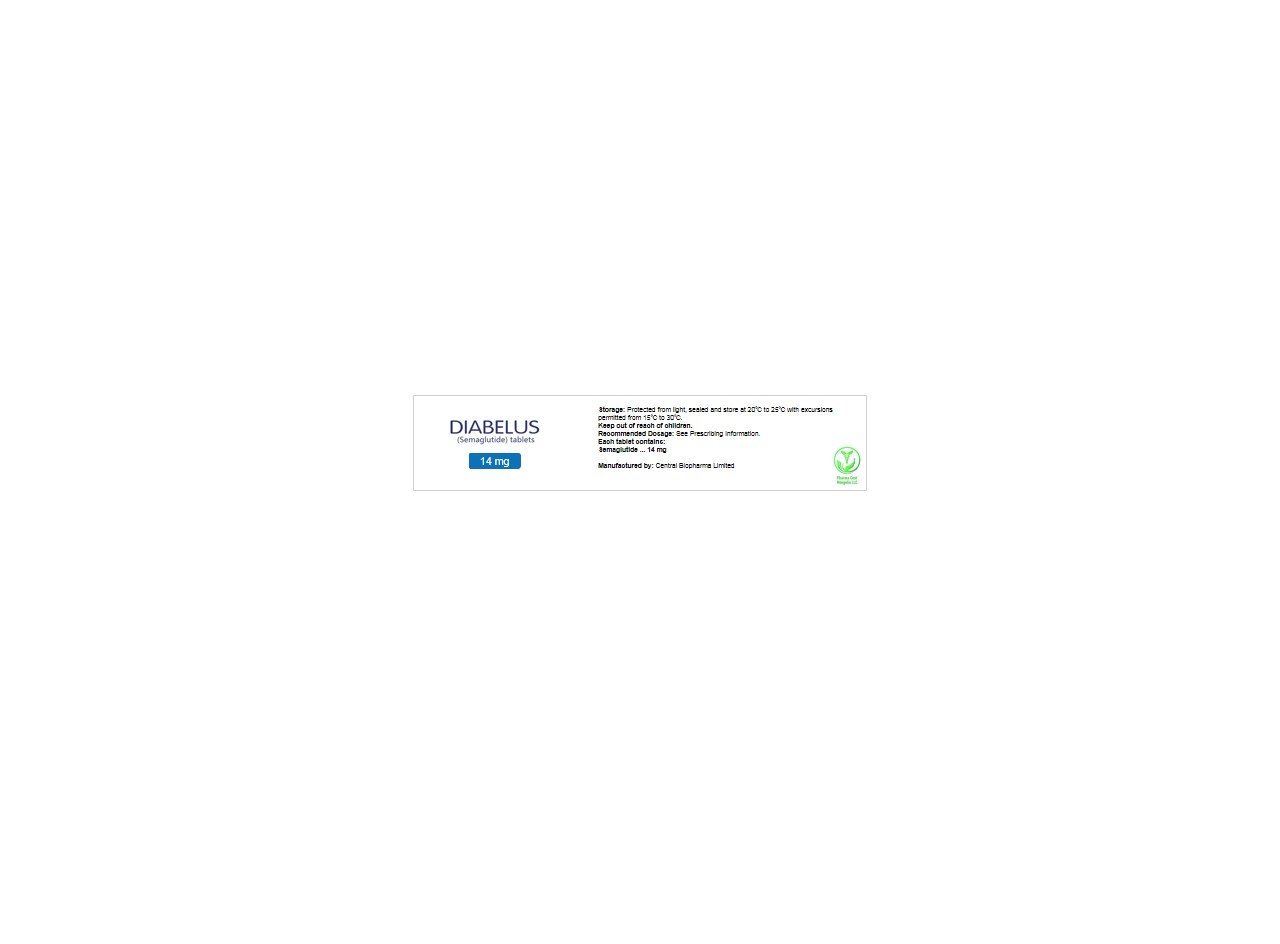
semaglutide includes the following strengths: 3 mg, 7 mg, and 14 mg.
Recommend the following semaglutide dosage to reduce the risk of gastrointestinal adverse reactions:
- Starting Dosage (Initiation Phase) (Days 1 to 30): The recommended starting dosage is 3 mg orally once daily (this dosage is not effective for glycemic control).
- Escalation and Maintenance Dosage (Days 31 and beyond):
o Days 31 to 60: Increase the dosage to 7 mg orally once daily.
o On Day 61 or thereafter, if:
- No additional glycemic control is needed, maintain the dosage at 7 mg orally once daily.
- Additional glycemic control is needed, increase the dosage to 14 mg orally once daily.
3 DOSAGE FORMS AND STRENGTHS
semaglutide tablets are available as: 3 mg, 7 mg, and 14 mg.
4 CONTRAINDICATIONS
semaglutide is contraindicated in patients with:
- A personal or family history of medullary thyroid carcinoma (MTC) or in patients with Multiple Endocrine Neoplasia syndrome type 2 (MEN 2).
- A prior serious hypersensitivity reaction to semaglutide or to any of the excipients in semaglutide. Serious hypersensitivity reactions including anaphylaxis and angioedema have been reported with semaglutide.
5 WARNINGS AND PRECAUTIONS
5.1 Risk of Thyroid C-Cell Tumors
In mice and rats, semaglutide caused a dose-dependent and treatment-duration-dependent increase in the incidence of thyroid C-cell tumors (adenomas and carcinomas) after lifetime exposure at clinically relevant plasma exposures. It is unknown whether semaglutide causes thyroid C-cell tumors, including medullary thyroid carcinoma (MTC), in humans as human relevance of semaglutide-induced rodent thyroid C-cell tumors has not been determined.
Cases of MTC in patients treated with liraglutide, another GLP-1 receptor agonist, have been reported in the postmarketing period; the data in these reports are insufficient to establish or exclude a causal relationship between MTC and GLP-1 receptor agonist use in humans.
semaglutide is contraindicated in patients with a personal or family history of MTC or in patients with MEN 2. Counsel patients regarding the potential risk for MTC with the use of semaglutide and inform them of symptoms of thyroid tumors (e.g., a mass in the neck, dysphagia, dyspnea, persistent hoarseness).
Routine monitoring of serum calcitonin or using thyroid ultrasound is of uncertain value for early detection of MTC in patients treated with semaglutide. Such monitoring may increase the risk of unnecessary procedures, due to the low test specificity for serum calcitonin and a high background incidence of thyroid disease. Significantly elevated serum calcitonin value may indicate MTC and patients with MTC usually have calcitonin values >50 ng/L. If serum calcitonin is measured and found to be elevated, the patient should be further evaluated. Patients with thyroid nodules noted on physical examination or neck imaging should also be further evaluated.
5.2 Acute Pancreatitis
Acute pancreatitis, including fatal and non-fatal hemorrhagic or necrotizing pancreatitis, has been observed in patients treated with GLP-1 receptor agonists, including semaglutide [see Adverse Reactions (6.1)].
After initiation of semaglutide, observe patients carefully for signs and symptoms of pancreatitis (including persistent severe abdominal pain, sometimes radiating to the back and which may or may not be accompanied by vomiting). If pancreatitis is suspected, discontinue semaglutide and initiate appropriate management.
5.3 Diabetic Retinopathy Complications
In a pooled analysis of glycemic control trials with semaglutide, patients reported diabetic retinopathy related adverse reactions during the trial (4.2% with semaglutide and 3.8% with comparator).
In a 2-year cardiovascular outcomes trial with semaglutide injection involving patients with type 2 diabetes mellitus and high cardiovascular risk, diabetic retinopathy complications (which was a 4-component adjudicated endpoint) occurred in patients treated with semaglutide injection (3%) compared to placebo (1.8%). The absolute risk increase for diabetic retinopathy complications was larger among patients with a history of diabetic retinopathy at baseline (semaglutide injection 8.2%, placebo 5.2%) than among patients without a known history of diabetic retinopathy (semaglutide injection 0.7%, placebo 0.4%).
Rapid improvement in glucose control has been associated with a temporary worsening of diabetic retinopathy. The effect of long-term glycemic control with semaglutide on diabetic retinopathy complications has not been studied. Patients with a history of diabetic retinopathy should be monitored for progression of diabetic retinopathy.
5.4 Hypoglycemia with Concomitant Use of Insulin Secretagogues or Insulin
Patients receiving semaglutide in combination with an insulin secretagogue (e.g., sulfonylurea) or insulin may have an increased risk of hypoglycemia, including severe hypoglycemia.
The risk of hypoglycemia may be lowered by a reduction in the dosage of sulfonylurea (or other concomitantly administered insulin secretagogue) or insulin. Inform patients using these concomitant medications of the risk of hypoglycemia and educate them on the signs and symptoms of hypoglycemia.
5.5 Acute Kidney Injury
There have been postmarketing reports of acute kidney injury and worsening of chronic renal failure, which may sometimes require hemodialysis, in patients treated with GLP-1 receptor agonists, including semaglutide. Some of these events have been reported in patients without known underlying renal disease. A majority of the reported events occurred in patients who had experienced nausea, vomiting, diarrhea, or dehydration.
Monitor renal function when initiating or escalating doses of semaglutide in patients reporting severe adverse gastrointestinal reactions.
5.6 Severe Gastrointestinal Adverse Reactions
Use of semaglutide has been associated with gastrointestinal adverse reactions, sometimes severe. In semaglutide clinical trials, severe gastrointestinal adverse reactions were reported more frequently among patients receiving semaglutide (7 mg 0.6%, 14 mg 2%) than placebo (0.3%).
semaglutide is not recommended in patients with severe gastroparesis.
5.7 Hypersensitivity Reactions
Serious hypersensitivity reactions (e.g., anaphylaxis, angioedema) have been reported in patients treated with semaglutide. If hypersensitivity reactions occur, discontinue use of semaglutide; treat promptly per standard of care, and monitor until signs and symptoms resolve. semaglutide is contraindicated in patients with a prior serious hypersensitivity reaction to semaglutide or to any of the excipients in semaglutide.
Anaphylaxis and angioedema have been reported with GLP-1 receptor agonists. Use caution in a patient with a history of angioedema or anaphylaxis with another GLP-1 receptor agonist because it is unknown whether such patients will be predisposed to anaphylaxis with semaglutide.
5.8 Acute Gallbladder Disease
Acute events of gallbladder disease such as cholelithiasis or cholecystitis have been reported in GLP-1 receptor agonist trials and postmarketing. In placebo-controlled trials, cholelithiasis was reported in 1% of patients treated with semaglutide 7 mg. Cholelithiasis was not reported in semaglutide 14 mg or placebo-treated patients. If cholelithiasis or cholecystitis is suspected, gallbladder studies and appropriate clinical follow-up are indicated.
5.9 Pulmonary Aspiration During General Anesthesia or Deep Sedation
semaglutide delays gastric emptying. There have been rare postmarketing reports of pulmonary aspiration in patients receiving GLP-1 receptor agonists undergoing elective surgeries or procedures requiring general anesthesia or deep sedation who had residual gastric contents despite reported adherence to preoperative fasting recommendations.
Available data are insufficient to inform recommendations to mitigate the risk of pulmonary aspiration during general anesthesia or deep sedation in patients taking semaglutide, including whether modifying preoperative fasting recommendations or temporarily discontinuing semaglutide could reduce the incidence of retained gastric contents. Instruct patients to inform healthcare providers prior to any planned surgeries or procedures if they are taking semaglutide.
6 ADVERSE REACTIONS
The following serious adverse reactions are described below or elsewhere in the prescribing information:
- Risk of Thyroid C-cell Tumors
- Acute Pancreatitis
- Diabetic Retinopathy Complications
- Hypoglycemia with Concomitant Use of Insulin Secretagogues or Insulin
- Acute Kidney Injury
- Severe Gastrointestinal Adverse Reactions
- Hypersensitivity Reactions
- Acute Gallbladder Disease
- Pulmonary Aspiration During General Anesthesia or Deep Sedation
7 DRUG INTERACTIONS
7.1 Concomitant Use with an Insulin Secretagogue (e.g., Sulfonylurea) or with Insulin
semaglutide stimulates insulin release in the presence of elevated blood glucose concentrations. Patients receiving semaglutide in combination with an insulin secretagogue (e.g., sulfonylurea) or insulin may have an increased risk of hypoglycemia, including severe hypoglycemia.
When initiating semaglutide, consider reducing the dose of concomitantly administered insulin secretagogue (such as sulfonylureas) or insulin to reduce the risk of hypoglycemia.
7.2 Other Oral Drugs
semaglutide causes a delay of gastric emptying, and thereby has the potential to impact the absorption of other oral drugs. Levothyroxine exposure was increased 33% (90% CI: 125-142) when administered with semaglutide in a drug interaction study.
When co-administering semaglutide with other oral drugs that have a narrow therapeutic index or that require clinical monitoring, consider increased clinical or laboratory monitoring.
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
Available data with semaglutide use in pregnant women are insufficient to evaluate for a drug-associated risk of major birth defects, miscarriage or other adverse maternal or fetal outcomes. There are clinical considerations regarding the risks of poorly controlled diabetes in pregnancy (see Clinical Considerations). Based on animal reproduction studies, there may be potential risks to the fetus from exposure to semaglutide during pregnancy. semaglutide should be used during pregnancy only if the potential benefit justifies the potential risk to the fetus.
In pregnant rats administered semaglutide during organogenesis, embryofetal mortality, structural abnormalities and alterations to growth occurred at maternal exposures below the maximum recommended human dose (MRHD) based on AUC. In rabbits and cynomolgus monkeys administered semaglutide during organogenesis, early pregnancy losses and structural abnormalities were observed at exposure below the MRHD (rabbit) and ≥10-fold the MRHD (monkey). These findings coincided with a marked maternal body weight loss in both animal species (see Data).
The estimated background risk of major birth defects is 6–10% in women with pre-gestational diabetes with an HbA1c >7 and has been reported to be as high as 20–25% in women with a HbA1c >10. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2-4% and 15-20%, respectively.
Clinical Considerations
Disease Associated Maternal and Fetal Risk: Poorly controlled diabetes during pregnancy increases the maternal risk for diabetic ketoacidosis, pre- eclampsia, spontaneous abortions, preterm delivery, and delivery complications. Poorly controlled diabetes increases the fetal risk for major birth defects, stillbirth, and macrosomia related morbidity.
Data
Animal Data: In a combined fertility and embryofetal development study in rats, subcutaneous doses of 0.01, 0.03 and 0.09 mg/kg/day (0.2-, 0.7-, and 2.1-fold the MRHD) were administered to males for 4 weeks prior to and throughout mating and to females for 2 weeks prior to mating, and throughout organogenesis to Gestation Day 17. In parental animals, pharmacologically mediated reductions in body weight gain and food consumption were observed at all dose levels. In the offspring, reduced growth and fetuses with visceral (heart blood vessels) and skeletal (cranial bones, vertebra, ribs) abnormalities were observed at the human exposure.
In an embryofetal development study in pregnant rabbits, subcutaneous doses of 0.0010, 0.0025 or 0.0075 mg/kg/day (0.06-, 0.6-, and 4.4-fold the MRHD) were administered throughout organogenesis from Gestation Day 6 to 19. Pharmacologically mediated reductions in maternal body weight gain and food consumption were observed at all dose levels. Early pregnancy losses and increased incidences of minor visceral (kidney, liver) and skeletal (sternebra) fetal abnormalities were observed at ≥0.0025 mg/kg/day, at clinically relevant exposures.
In an embryofetal development study in pregnant cynomolgus monkeys, subcutaneous doses of 0.015, 0.075, and 0.15 mg/kg twice weekly (1.9-, 9.9-, and 29-fold the MRHD) were administered throughout organogenesis, from Gestation Day 16 to 50. Pharmacologically mediated, marked initial maternal body weight loss and reductions in body weight gain and food consumption coincided with the occurrence of sporadic abnormalities (vertebra, sternebra, ribs) at ≥0.075 mg/kg twice weekly (>9X human exposure).
In a pre- and postnatal development study in pregnant cynomolgus monkeys, subcutaneous doses of 0.015, 0.075, and 0.15 mg/kg twice weekly (1.3-, 6.4-, and 14-fold the MRHD) were administered from Gestation Day 16 to
- Pharmacologically mediated marked initial maternal body weight loss and reductions in body weight gain and food consumption coincided with an increase in early pregnancy losses and led to delivery of slightly smaller offspring at ≥0.075 mg/kg twice weekly (>6X human exposure).
Salcaprozate sodium (SNAC), an absorption enhancer in semaglutide, crosses the placenta and reaches fetal tissues in rats. In a pre- and postnatal development study in pregnant Sprague Dawley rats, SNAC was administered orally at 1,000 mg/kg/day (exposure levels were not measured) on Gestation Day 7 through lactation day 20. An increase in gestation length, an increase in the number of stillbirths and a decrease in pup viability were observed.
8.2 Lactation
Risk Summary
A clinical lactation study reported semaglutide concentrations below the lower limit of quantification in human breast milk. However, salcaprozate sodium (SNAC) and/or its metabolites are present in human milk. Since the activity of enzymes involved in SNAC clearance may be lower in infants compared to adults, higher SNAC plasma levels may occur in neonates and infants. Because of the unknown potential for serious adverse reactions in the breastfed infant due to the possible accumulation of SNAC, and because there are alternative formulations of semaglutide that do not contain SNAC that can be used during lactation, advise patients that breastfeeding is not recommended during treatment with semaglutide.
8.3 Females and Males of Reproductive Potential
Discontinue semaglutide in women at least 2 months before a planned pregnancy due to the long washout period for semaglutide.
8.4 Pediatric Use
The safety and effectiveness of semaglutide have not been established in pediatric patients.
8.5 Geriatric Use
In the pool of glycemic control trials, 1,229 (30%) semaglutide-treated patients were 65 years of age and over and 199 (5%) semaglutide-treated patients were 75 years of age and over [see Clinical Studies (14)]. In PIONEER 6, the cardiovascular outcomes trial, 891 (56%) semaglutide-treated patients were 65 years of age and over and 200 (13%) semaglutide-treated patients were 75 years of age and over.
No overall differences in safety or effectiveness for semaglutide have been observed between patients 65 years of age and older and younger adult patients.
8.6 Renal Impairment
The recommended dosage of semaglutide in patients with renal impairment is the same as those with normal renal function.
The safety and effectiveness of semaglutide was evaluated in a 26-week clinical study that included 324 patients with moderate renal impairment (eGFR 30 to 59 mL/min/1.73 m2). In patients with renal impairment including end-stage renal disease (ESRD), no clinically relevant change in semaglutide pharmacokinetics was observed.
8.7 Hepatic Impairment
The recommended dosage in patients with hepatic impairment is the same as those with normal hepatic function.
In a study in subjects with different degrees of hepatic impairment, no clinically relevant change in semaglutide pharmacokinetics was observed.
9 OVERDOSAGE
In the event of overdose, appropriate supportive treatment should be initiated according to the patient’s clinical signs and symptoms. A prolonged period of observation and treatment for these symptoms may be necessary, taking into account the long half-life of semaglutide of approximately 1 week.
10 DESCRIPTION
semaglutide tablets, for oral use, contain semaglutide, a GLP-1 receptor agonist. The peptide backbone is produced by yeast fermentation. The main protraction mechanism of semaglutide is albumin binding, facilitated by modification of position 26 lysine with a hydrophilic spacer and a C18 fatty di-acid. Furthermore, semaglutide is modified in position 8 to provide stabilization against degradation by the enzyme dipeptidyl-peptidase 4 (DPP-4). A minor modification was made in position 34 to ensure the attachment of only one fatty di-acid. The molecular formula is C187H291N45O59 and the molecular weight is 4113.58 g/mol.
Structural formula:
Semaglutide is a white to almost white hygroscopic powder. Each tablet of:
- semaglutide contains 3 mg, 7 mg or 14 mg of semaglutide and the following inactive ingredients: magnesium stearate, microcrystalline cellulose, povidone and salcaprozate sodium (SNAC).
11 MECHANISM OF ACTION
Semaglutide is a GLP-1 analogue with 94% sequence homology to human GLP-1. Semaglutide acts as a GLP-1 receptor agonist that selectively binds to and activates the GLP-1 receptor, the target for native GLP-1.
GLP-1 is a physiological hormone that has multiple actions on glucose, mediated by the GLP-1 receptors.
The principal mechanism of protraction resulting in the long half-life of semaglutide is albumin binding, which results in decreased renal clearance and protection from metabolic degradation. Furthermore, semaglutide is stabilized against degradation by the DPP-4 enzyme.
Semaglutide reduces blood glucose through a mechanism where it stimulates insulin secretion and lowers glucagon secretion, both in a glucose-dependent manner. Thus, when blood glucose is high, insulin secretion is stimulated and glucagon secretion is inhibited. The mechanism of blood glucose lowering also involves a minor delay in gastric emptying in the early postprandial phase.
12 HOW SUPPLIED/STORAGE AND HANDLING
How Supplied
Semaglutide tablets are available as oval-shaped, white to light yellow tablets in the following strengths: 3 mg, 7 mg, 14 mg.
Each bottle contains 30 tablets.
Storage and Handling
Store at 68°F to 77°F (20°C to 25°C); excursions permitted to 59°F to 86°F (15°C to 30°C) [see USP Controlled Room Temperature]. Store and dispense in the original bottle.
Store tablet in the original bottle until use to protect tablets from moisture. Store product in a dry place away from moisture.
13 PATIENT COUNSELING INFORMATION
Advise the patient to read the approved labeling (Medication Guide).
Risk of Thyroid C-cell Tumors
Inform patients that semaglutide causes thyroid C-cell tumors in rodents and that the human relevance of this finding has not been determined. Counsel patients to report symptoms of thyroid tumors (e.g., a lump in the neck, hoarseness, dysphagia, or dyspnea) to their physician.
Acute Pancreatitis
Inform patients of the potential risk for acute pancreatitis and its symptoms: severe abdominal pain that may radiate to the back, and which may or may not be accompanied by vomiting. Instruct patients to discontinue semaglutide promptly and contact their physician if pancreatitis is suspected.
Diabetic Retinopathy Complications
Inform patients to contact their physician if changes in vision are experienced during treatment with semaglutide.
Hypoglycemia with Concomitant Use of Insulin Secretagogues or Insulin
Inform patients that the risk of hypoglycemia is increased when semaglutide is used with an insulin secretagogue (such as a sulfonylurea) or insulin. Educate patients on the signs and symptoms of hypoglycemia.
Dehydration and Renal Failure
Advise patients treated with semaglutide of the potential risk of dehydration due to gastrointestinal adverse reactions and take precautions to avoid fluid depletion. Inform patients of the potential risk for worsening renal function and explain the associated signs and symptoms of renal impairment, as well as the possibility of dialysis as a medical intervention if renal failure occurs.
Severe Gastrointestinal Adverse Reactions
Inform patients of the potential risk of severe gastrointestinal adverse reactions. Instruct patients to contact their healthcare provider if they have severe or persistent gastrointestinal symptoms.
Hypersensitivity Reactions
Inform patients that serious hypersensitivity reactions have been reported during postmarketing use of semaglutide. Advise patients on the symptoms of hypersensitivity reactions and instruct them to stop taking semaglutide and seek medical advice promptly if such symptoms occur.
Acute Gallbladder Disease
Inform patients of the potential risk for cholelithiasis or cholecystitis. Instruct patients to contact their physician if cholelithiasis or cholecystitis is suspected for appropriate clinical follow-up.
Pulmonary Aspiration During General Anesthesia or Deep Sedation:
Inform patients that semaglutide may cause their stomach to empty more slowly which may lead to complications with anesthesia or deep sedation during planned surgeries or procedures. Instruct patients to inform healthcare providers prior to any planned surgeries or procedures if they are taking semaglutide.
Pregnancy
Advise a pregnant woman of the potential risk to a fetus. Advise women to inform their healthcare provider if they are pregnant or intend to become pregnant.
Lactation
semaglutide passes into breast milk, and it is not known how it affects your baby. Advise females not to breastfeed during treatment with semaglutide.
Females and Males of Reproductive Potential
Discontinue semaglutide at least 2 months before a planned pregnancy due to the long washout period for semaglutide.
14 EXPIRY DATE
24 months after the date of manufacturing.
{kind=link}
{kind=link}